Biologie Systémique et Computationnelle
La recherche pharmaceutique passe en mode numérique
Date:
Date:
Mis à jour le 03/02/2023
« Cette thèse Cifre*, c’est finalement une concrétisation d’une action exploratoire en biologie des systèmes que nous avions menée chez Inria dès 2001, s’amuse François Fages, directeur de l’équipe-projet Inria Lifeware. La preuve que le dispositif porte ses fruits ! » Le flambeau sur ce sujet a en effet été repris en 2017 par Jeremy Grignard, au cours d’un stage de recherche dans l’équipe de François Fages, dans le cadre de son cursus d’ingénieur en informatique et big data à l’École centrale d’électronique. Puis porté jusqu’à une thèse Cifre, alliant deux partenaires, académique et industriel. Si le centre Inria de Saclay a donné son accord pour occuper le rôle du premier, Jeremy Grignard doit alors trouver le second et démarche différentes sociétés, dont des industries pharmaceutiques, en présentant l’idée de sa thèse : l’apprentissage automatique de modèles à partir d’expériences et de données temporelles.
Servier lui ouvre ses portes, car derrière l’idée de la modélisation se cache un enjeu de taille pour l’industrie pharmaceutique : mieux comprendre les processus biochimiques et les voies de signalisation impliquant une cible thérapeutique dérégulée dans une maladie pour mieux identifier les candidats-médicaments qui pourront la moduler. Et économiser ainsi du temps et de l’argent. Car plus de 58% des candidats-médicaments échouent en phase III (essais sur de larges cohortes de patients présentant la maladie à traiter). Des années et des milliards d’euros d’investissement sont ainsi perdus.
« Dans nos besoins, il y avait une grosse part d’investigation, qui se prêtait bien à la thèse, résume Thierry Dorval, directeur du département sciences et gestion des données chez Servier. Et nous ne manquions pas de données expérimentales pour les recherches ! » La question était bien de savoir comment leur donner du sens et les exploiter au mieux.
Jeremy Grignard commence par un cas concret : des molécules développées par Servier ont un effet au niveau biochimique sur une protéine impliquée dans un processus particulier (la tyrosination des microtubules)… mais cette activité ne se retrouve pas au niveau cellulaire. Et les chercheurs ne comprenaient pas pourquoi. Le jeune ingénieur met alors au point un modèle mathématique expert à partir des données du laboratoire… et celui-ci fournit l’explication : le processus biologique repose sur deux réactions ; n’en activer qu’une ne suffit donc pas !
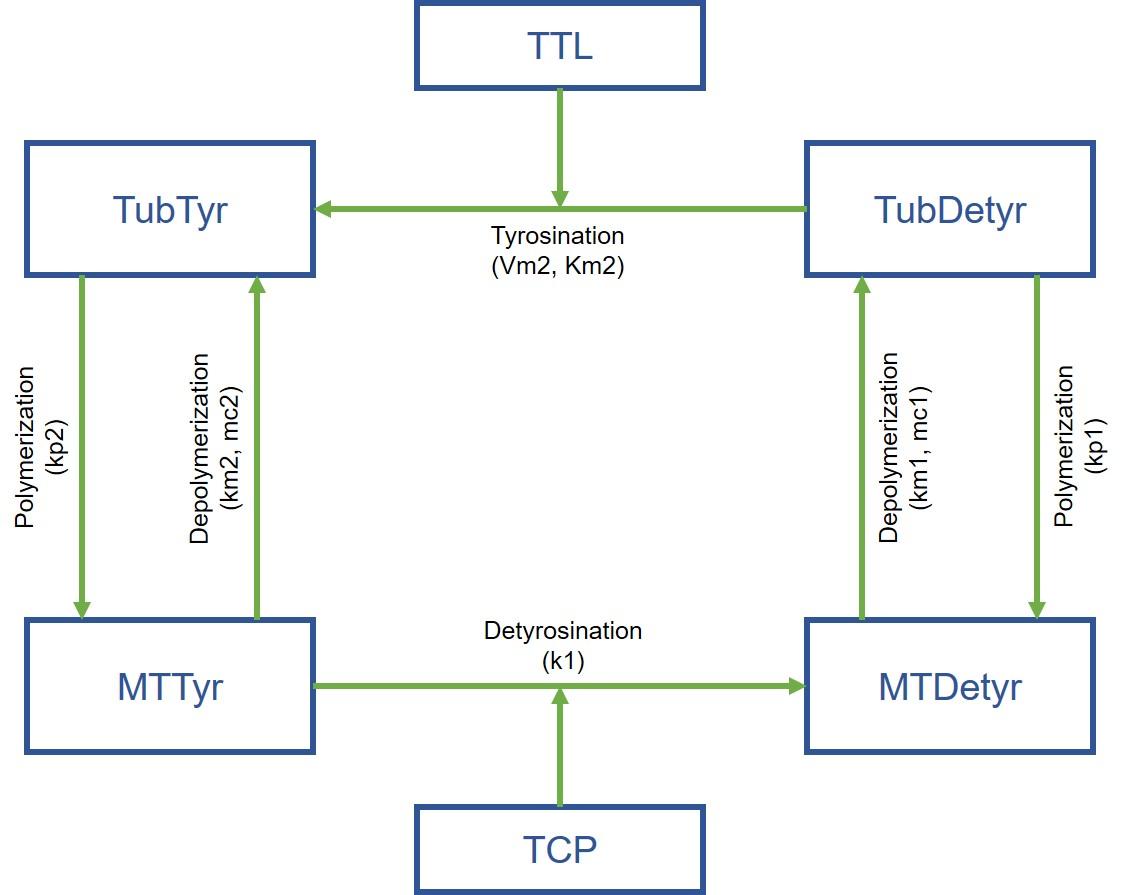
Le modèle a en outre mis en évidence une autre protéine, qui, en étant cette fois inhibée, permet d’aboutir au résultat souhaité. Une nouvelle piste pour les chercheurs. « L’idée est à présent de développer ce type de modèle pour chaque projet thérapeutique, annonce Thierry Dorval. Cela implique de se pencher sur l’apprentissage automatique de modèles… et ce sera le sujet d’une deuxième thèse à venir. »
Si ce thème était en effet au départ au cœur de la thèse de Jeremy Grignard, cette dernière a légèrement évolué. « Nous nous sommes rendu compte que les données temporelles, sur lesquelles devaient se baser les recherches de Jeremy, n’étaient pas celles dont nous disposions le plus, poursuit le spécialiste de Servier. Les données d’imagerie étaient bien plus nombreuses. » La thèse de Jeremy Grignard a donc été renommée « Méthodes computationnelles pour améliorer les phases primaires de recherche de nouveaux médicaments » et le chercheur s’est penché sur l’exploitation des données d’imagerie. Il s’est attaché en particulier aux expériences de criblage phénotypiques.
Celles-ci consistent à colorer des cellules, puis, à l’aide de logiciels d’analyse d’images, à observer les modifications de leur apparence (leur phénotype) en réponse à une perturbation génétique ou à une molécule chimique. « Le problème réside dans le grand nombre de variables mesurées, qu’il faut pouvoir normaliser pour espérer ensuite lier une modification phénotypique avec l’activité d’une molécule ou d’une perturbation génétique », souligne Jeremy Grignard. Et pour cela, il a développé deux algorithmes permettant, justement, cette normalisation.

Leur application potentielle est énorme puisque Servier fait partie du consortium international JUMP-CP, qui rassemble une dizaine de grandes entreprises pharmaceutiques sous l’égide du Broad Institute du MIT et d’Harvard. Celui-ci s’est fixé pour mission de réaliser la plus grande expérience de criblage phénotypique en étudiant les effets de 120 000 molécules chimiques et de perturbations génétiques CRISPR/Cas9. Utiliser les algorithmes de normalisation mis au point par Jeremy Grignard, et en développer d’autres, permettra l’exploitation des données massives ainsi générées.
Restait la question : comment ? Réponse : grâce au graphe de connaissances. Cet outil mathématique permet d’intégrer des données très hétérogènes et de volumétrie très importante, ainsi que les relations qui les lient, à un logiciel capable d’exploiter l’ensemble. Celui développé par Jeremy Grignard s’appelle Pegasus : il contient aujourd’hui environ 46 millions de nœuds, c’est-à-dire de données, issues de la recherche interne à Servier ou du domaine public, ainsi que 331 millions de relations entre ces données.
Si de tels chiffres rendent l’outil presque abstrait, ses applications, elles, sont bien concrètes. Pegasus a par exemple déjà été utilisé pour sélectionner des oligonucléotides antisens (Asos), de petites séquences génétiques capables de moduler l’expression d’une protéine cible, dans une maladie rare : l’encéphalopathie épileptique. À l’avenir, l’objectif est de permettre aux chercheurs de Servier de repérer rapidement les Asos potentiels pour une cible thérapeutique donnée.
Pegasus permet également d’identifier les molécules développées par Servier chimiquement similaires à des molécules étudiées dans des publications scientifiques pour leur effet sur une cible thérapeutique. « Au lieu de cribler un grand nombre de molécules pour voir si elles agissent sur la cible, ce qui aboutit, en moyenne, à un taux de succès de 1%, nous pourrons nous baser sur les relations de similarités présentes dans Pegasus pour cribler directement les molécules les plus intéressantes. Et ainsi augmenter significativement le taux de succès et la qualité des molécules identifiées », s’enthousiasme Jeremy Grignard.
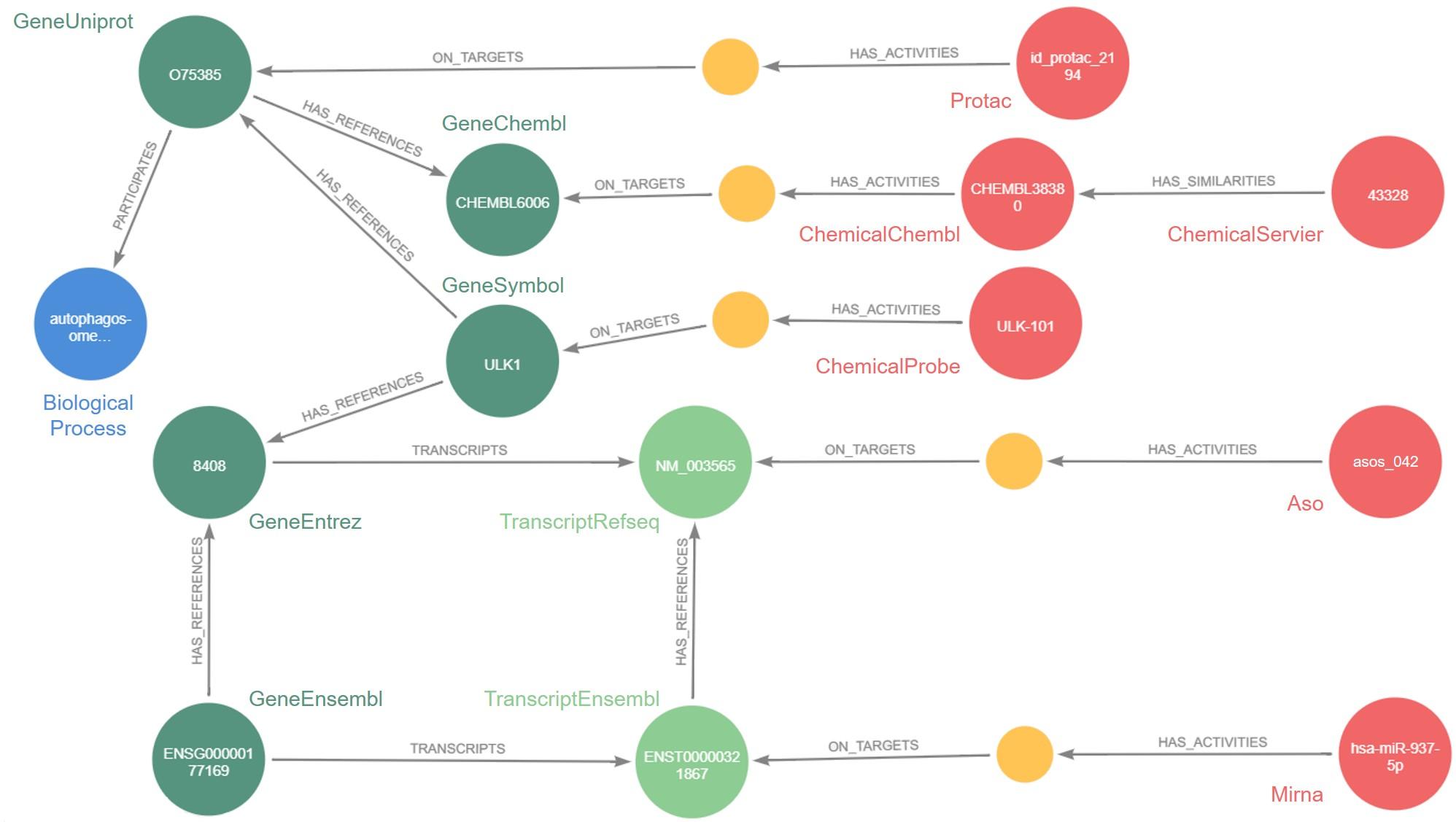
Au final, les trois outils sont parfaitement complémentaires : Pegasus est nourri des données issues des modèles mathématiques et de celles des expériences de criblage phénotypique que les algorithmes permettent de normaliser. La boucle est bouclée… ou presque. Car les stocks de données sont énormes et Pegasus ainsi que les applications industrielles associées sont amenés à évoluer.
C’est d’ailleurs pour cela que Jeremy Grignard a été embauché immédiatement après ses trois ans de thèse, en mars 2022, au sein de l’Institut de recherches Servier, en tant que Data Scientist. « La thèse Cifre permet de répondre à des questions très concrètes que se pose un industriel en développant des approches théoriques que nous n’aurions pas envisagées nous-mêmes, estime François Fages. En outre, les outils développés peuvent nous ouvrir de nouvelles perspectives : nous avons quelques projets de recherche pour lesquels Pegasus pourrait être très intéressant. » La thèse Cifre a fait naître une collaboration qui n’a peut-être pas dit son dernier mot…
* Convention industrielle de formation par la recherche.